Article Text

Abstract
Background It has been hypothesised that an upregulation of the neuropeptide substance P (SP) and its preferred receptor, the neurokinin-1 receptor (NK-1 R), is a causative factor in inducing tenocyte hypercellularity, a characteristic of tendinosis, through both proliferative and antiapoptotic stimuli. We have demonstrated earlier that SP stimulates proliferation of human tenocytes in culture.
Aim The aim of this study was to investigate whether SP can mediate an antiapoptotic effect in tumour necrosis factor-α (TNF-α)-induced apoptosis of human tenocytes in vitro.
Results A majority (approximately 75%) of tenocytes in culture were immunopositive for TNF Receptor-1 and TNF Receptor-2. Exposure of the cells to TNF-α significantly decreased cell viability, as shown with crystal violet staining. TNF-α furthermore significantly increased the amount of caspase-10 and caspase-3 mRNA, as well as both BID and cleaved-poly ADP ribosome polymerase (c-PARP) protein. Incubation of SP together with TNF-α resulted in a decreased amount of BID and c-PARP, and in a reduced lactate dehydrogenase release, as compared to incubation with TNF-α alone. The SP effect was blocked with a NK-1 R inhibitor.
Discussion This study shows that SP, through stimulation of the NK-1 R, has the ability to reduce TNF-α-induced apoptosis of human tenocytes. Considering that SP has previously been shown to stimulate tenocyte proliferation, the study confirms SP as a potent regulator of cell-turnover in tendon tissue, capable of stimulating hypercellularity through different mechanisms. This gives further support for the theory that the upregulated amount of SP seen in tendinosis could contribute to hypercellularity.
- Achilles Tendon
- Tendons
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
The pathogenesis of tendinosis, that is, chronic tendon pain accompanied by structural tissue changes, has not been fully elucidated. It has been suggested that biochemical mediators produced by the primary tendon cells (tenocytes) in response to stress may play a causative role1 ,2; a theory supported by studies showing that tenocytes express enzymes related to synthesis of classically neuronal substances, such as acetylcholine and catecholamines.3–5 A neuropeptide that has gained interest in the development of tendinosis is substance P (SP).
SP has been shown to elicit diverse cellular responses, ranging from its ability to transmit pain and mediate inflammatory responses, to stimulating angiogenesis.6 SP belongs to the tachykinin neuropeptide family, and its preferred receptor, the neurokinin-1 receptor (NK-1 R), belongs to the tachykinin subfamily of G protein-coupled receptors (GPCR).7 The ability of GPCRs to transduce signals that stimulate cellular proliferation is well established.8
It has been shown that tenocytes of the human Achilles tendon express mRNA for SP as well as its receptor NK-1 R on both mRNA and protein level.9 It has also been suggested that elevated levels of SP, due to mechanical overload, leads to proliferation of the tenocytes through an autocrine loop involving a NK-1 R specific pathway, implicating SP to have a role in the development of histopathological features seen in tendinosis, such as hypercellularity and angiogenesis.10–12 It should, in the context of SP elevation, be mentioned that increased SP has also been seen in tendinosis as compared to normal tendon tissue.13–15
Another major observation in biopsies from tendinosis tissue is the high amount of apoptotic cells.16 The causes of this excessive apoptosis, and the possible role of SP and NK-1 R in this process, are still poorly defined. It has previously been suggested that the cytokine TNF-α has an effect on the level of apoptosis in tenocytes.17
During mechanotransduction, cytokines play a pivotal role as signalling substances. Since tenocytes adapt to loading through mechanotransduction,18 it is not surprising that changes in the cytokine-signalling pathway, like for TNF-α,19 are implicated in tendinosis. TNF-α and its receptors, tumour necrosis factor receptor 1 and tumour necrosis factor receptor 2 (TNF-R1 and TNF-R2), have been identified in the human Achilles tendon19 as well as in cultured tenocytes.20
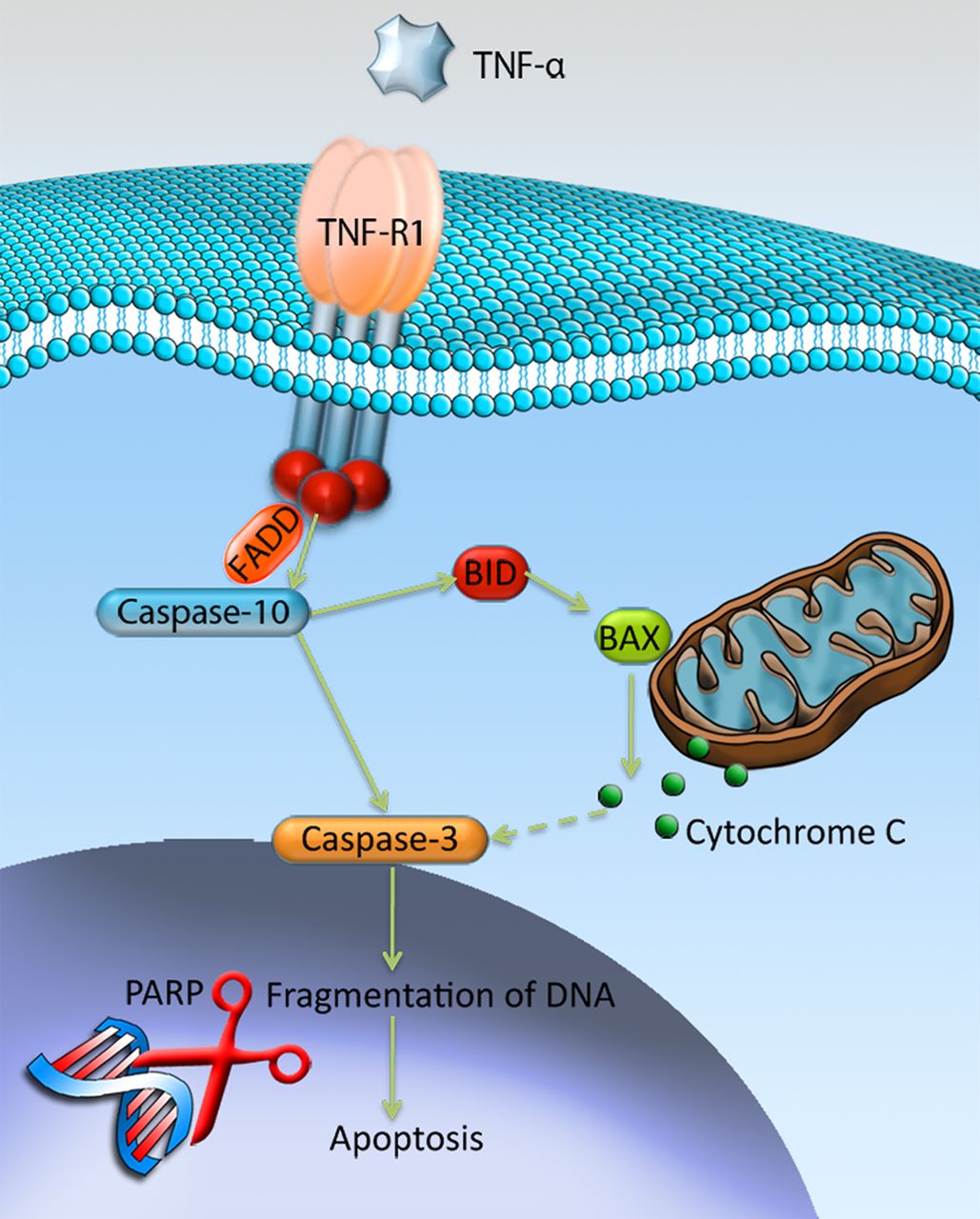
TNF-R1 has the ability to signal cell death via its cytoplasmic death domain by recruiting the adaptor protein Fas-Associated protein with Death Domain (FADD), which subsequently cleaves and activates caspase-10.21 ,22 Cleaved/activated caspase-10 has the ability to activate downstream caspases, such as caspase-3, by directly cleaving them.23 In addition, cleaved caspase-10 has the ability to activate downstream caspases indirectly by cleaving the Bcl-2 family protein BH3 interacting-domain death agonist (BID), which instead targets the mitochondria and induces release of cytochrome C.23 ,24 At the end stage of apoptosis, when the DNA is fragmentised, the cleaved poly ADP ribosome polymerase (c-PARP) is involved. PARP is a protein that is involved in DNA repair, but the repairing function is inactivated by its cleavage, which thereby contributes to apoptosis and c-PARP is thus an established marker of cells undergoing apoptosis.25 See figure 1 for an overview.

Mechanisms of tumour necrosis factor-α (TNF-α)-induced apoptosis through the TNF-R1. Binding of TNF-α to TNF-R1 results in activation of caspase-10 through recruitment of the Fas-Associated protein with Death Domain (FADD). Activated caspase-10 has the ability to activate caspase-3, either directly by cleavage, or indirectly by activating the Bcl-2 family protein BID that through release of cytochrome-c will activate caspase-3. Ultimately, activated caspase-3 will result in fragmentation of DNA, that is, apoptosis, with the involvement of cleaved-poly ADP ribosome polymerase (PARP). Illustration by Gustav Andersson (copyright with artist).
SP has previously been shown to protect cells from apoptotic inducing agents.26–28 It does so by binding to the NK-1 R, causing phosphorylation and activation of the protein Akt.27 The phosphorylated form of Akt has been shown to play a pivotal role in different cell types by its distinct signalling network that modulates cellular proliferation, survival and apoptosis.29 Activated Akt has the ability to promote cellular survival by inactivating key mediators of the pro-apoptotic pathway, such as BID.29 An antiapoptotic effect of SP through phosphorylation of Akt has recently been observed in tenocytes after anti-Fas exposure.28
We hypothesise that TNF-α can induce apoptosis in human tenocytes and that this effect may be inhibited by SP. To test this hypothesis, we use a primary human Achilles tendon cell culture model, in which the phenotype of the tenocytes has recently been validated.11 ,30 The specific aims of the current study were to use this in vitro model to (1) study the expression of the receptors for TNF-α in tenocytes, (2) test whether stimulation with exogenous TNF-α induces apoptosis in tenocytes, and if so, through what mechanisms and (3) test whether exogenous SP can reduce the apoptosis induced by TNF-α and if so, whether this is a NK-1 R-mediated effect.
Material and methods
Ethical statement
The study was performed according to the principles of the Declaration of Helsinki. All tissue donors signed a written consent form.
Reagents
For experimental trials with TNF-α cells were incubated with two different concentrations (50 and 500 ng/mL) of TNF-α (PeproTech, Rocky Hill, New Jersey, USA; code: 300-01A). For experimental trials with SP, the cells were incubated in SP peptide (Calbiochem, San Diego, California, USA; code: 05-23-0600) at a concentration of 10−7 M, based on earlier studies11 30 min prior to TNF-α. An NK-1 R antagonist, L-733 060 hydrochloride (Tocris, Minneapolis, Minnesota, USA; code:1145), was used in the concentration of 10−6 M, based on an earlier study.11 The NK-1 R antagonist was incubated 30 min prior to exposure with SP.
Harvesting and culturing of human Achilles tendon cells
Samples of human Achilles tendons were harvested from the midportion of the tendon in participants who had no history of tendon pain and no structural changes seen at ultrasound examination. In this study, a total of three biopsies were used from two males and one female with an age span of 23–29 years at the time of biopsy collection, and all with the same ethnicity (Scandinavian). All experiments were performed at least three times, on cells from at least two different biopsies, to confirm the results. No differences in behaviour or phenotype of cells from the different participants were noticed during culture, nor were any differences in response to treatments observed.
The human Achilles tendon cell culture was performed as previously reported by our laboratory.11 ,30 In all experiments, the concentration of serum was reduced from 10% to 1% to eliminate any unwanted effects of the serum.
We have, in previous studies,11 ,30 confirmed that the cultured isolated human tendon cells are tenocytes, as they express scleraxis, tenomodulin and also collagen type I to a higher extent than collagen type III, in passages used for experiments.
Immunofluorescence
Fixation of cells was performed with 2% paraformaldehyde for 15 min and thereafter washed with phosphate-buffered saline (PBS) and blocked with 5% normal donkey serum for 15 min at room temperature. The cells were then incubated with a TNF-R1 goat antibody at a concentration of 1:50 (Santa-Cruz, California, USA; code: sc-1070) or a TNF-R2 goat antibody at a concentration of 1:50 (Santa-Cruz; code: sc-1074) for 60 min at 37°C. Afterwards cells were washed, blocked with 5% normal donkey serum and incubated with fluorescein isothiocyanate (FITC)-conjugated donkey anti-goat secondary antibody at a concentration of 1:300 (Jackson immunoresearch, Baltimore Pike, Pennsylvania, USA; code: 705-095-147) for 30 min at 37°C. After washing, slides were mounted using Vectashield Hard Set Medium with 4,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, California, USA; code: H-1500). The cells were examined using flourescence microscope, Zeiss Axioskop 2 Plus microscope with epifluorescence and an Olympus DP70 digital camera.
Lactate dehydrogenase assay
Cells were treated with TNF-α and the supernatant, used for analysing, was collected 48 h after the start of the experiment. Following the manufacture’s protocol (Roche Applied Science, Mannheim, Germany; code: 11 644 793 001) 100 μL of the supernatant was transferred to a 96-well plate together with one volume of reaction solution. Following incubation at room temperature for 30 min the absorbance was read at 490 nm. Before analysing the effects of TNF-α, the effects of SP and the NK-1 R inhibitor alone on the cells were checked for and no significant difference as compared to the control was seen.
Crystal violet (Cell viability assay)
Cells were analysed 48 h after treatment. Non-adherent cells were removed by washing with PBS before cells were fixed in 1% glutaraldehyde for 30 min. The cells were subsequently stained for 30 min in 0.1% crystal violet (Sigma, Saint Louis, Missouri, USA; code: C3886) and thereafter washed in water. After air-drying, the staining was dissolved in 30% methanol and 10% acetic acid and transferred to a 96-well plate before the absorbance was read at 590 nm.
RNA isolation and RT-quantitative PCR
The RNA extraction was prepared using RNAeasy-kit (Qiagen, Hilden, Germany; code: 74106) following the manufacture’s protocol. The RNA was thereafter reversed transcribed into cDNA by diluting the sample in a mixture containing 10× RT buffer, 25× dNTP, 10× RT random primers and 10× multiscribe RTase (Applied Biosystems, Foster city, California, USA; code: 4368813).
The real-time quantitative PCR (RT-qPCR) was performed using TaqMan fast universal PCR mastermix (Applied Biosystems; code: 4352042) and probes for β-actin (Applied biosystems; code: 4333762F), caspase-3 (Applied biosystems; code: Hs00234387) and caspase-10 (Applied biosystems; code: Hs01017899).
Western blot
Cells were lysed in RIPA buffer supplemented with protease inhibitor 1:200 (all from Sigma Aldrich). Protein determination was performed with protein Assay Dye Reagent Concentrate (Bio-Rad, Hercules, California, USA; code: 500-0006). Samples were boiled and thereafter separated on SDS-PAGE under reducing conditions of 160 V for 45 min. The separated proteins were transferred to polyvinylidene flouride transfer membrane (PVDF; Santa Cruz; code: sc-3723) at 100 V for 1 h. Thereafter, the membrane was blocked in blocking buffer, TBS-T, supplemented with 5% dried milk and thereafter incubated, over night at 4°C, with primary antibodies for BID (Cell Signaling, Danvers, Massachusetts, USA; code: 2002), cleaved PARP (c-PARP; Cell Signaling; code: 9541) and β-actin (Cell Signaling; code: 4967), all at a concentration of 1:1000. Cell lysates from jurkat apoptosis cells (Cell Signaling; code: 2043) were used as positive controls of apoptosis. After washing, incubation was performed with HRP-conjugated secondary antibody at a concentration of 1:2000 (Cell Signaling; code: 7074) for 1 h at room temperature. The membranes were washed and treated with prime western blotting detection reagent (GE healthcare, Little Chalfont, Buckinghamshire, UK; code: RPN2232) before being visualised on high-performance chemiluminescence film (GE healthcare; code: 28-9068-38).
Reblotting of the membrane with β-actin was performed after treatment with western blot stripping buffer (Thermo Scientific Rockford, Illinois, USA; code: 21059) at 37°C for 25 min to confirm equal loading.
Statistics
Triplets were used for the cytotoxicity assay, cell viability assay and RT-qPCR.
Data were analysed using Predictive Analytics SoftWare (PASW) statistics V.18 (18.0.0; SPSS Inc, Chicago, Illinois, USA). Statistical tests were analysed with one-way analysis of variance, followed by Bonferroni post hoc test. Significance was predetermined at p<0.05. All results were successfully repeated at least twice.
Results
Presence of TNF-R1 and TNF-R2 in human tendon cells in vitro
Marked immunoreactions for TNF-R1 (figure 2A) and TNF-R2 (figure 2B) were seen in the tenocytes. The majority (approximately 75%) of cells expressed both types of receptors, although TNF-R1 was over all more intensely expressed.
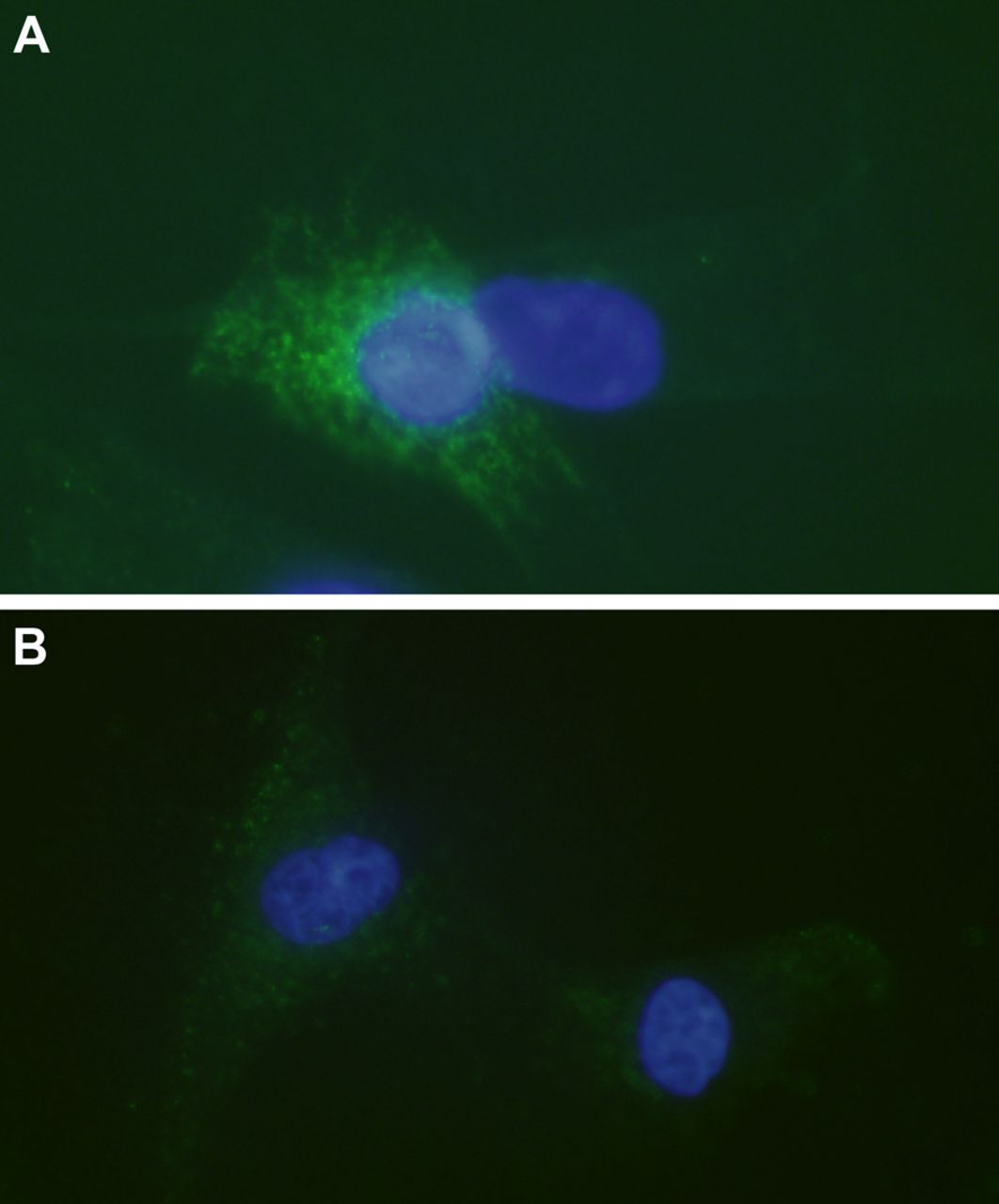
Tumour necrosis factor-α (TNF-α) receptors in cultured human tenocytes. Immunocytochemistry to confirm the presence of TNF-R1 (A) and TNF-R2 (B) in cultured human tenocytes (green; FITC). Cells are counterstained with DAPI (blue) to mark the nuclei (×600).
TNF-α decreases cell viability
There was a significant decrease in cell viability, as shown with crystal violet staining, in cells treated with 500 ng/mL as well as 50 ng/mL, of TNF-α as compared with control after 72 h of exposure (p<0.01, figure 3). The difference seen between cells treated with 50 and 500 ng/mL was not statistically significant, albeit was close to (p=0.07; figure 3).

Reduced cell viability after tumour necrosis factor-α (TNF-α) incubation. A significant decrease in cell viability of cultured human tenocytes was seen in cells treated with both 50 ng/mL of TNF-α and 500 ng/mL TNF-α, as compared to the control after 72 h of exposure (**p<0.01). The difference in cell viability seen between the treatments with different concentrations of TNF-α was not statistically significant (n.s.; p=0.07). Cell viability was measured with crystal violet staining (A590). Error bars indicate SD.
TNF-α activates caspases and BID at mRNA and protein level
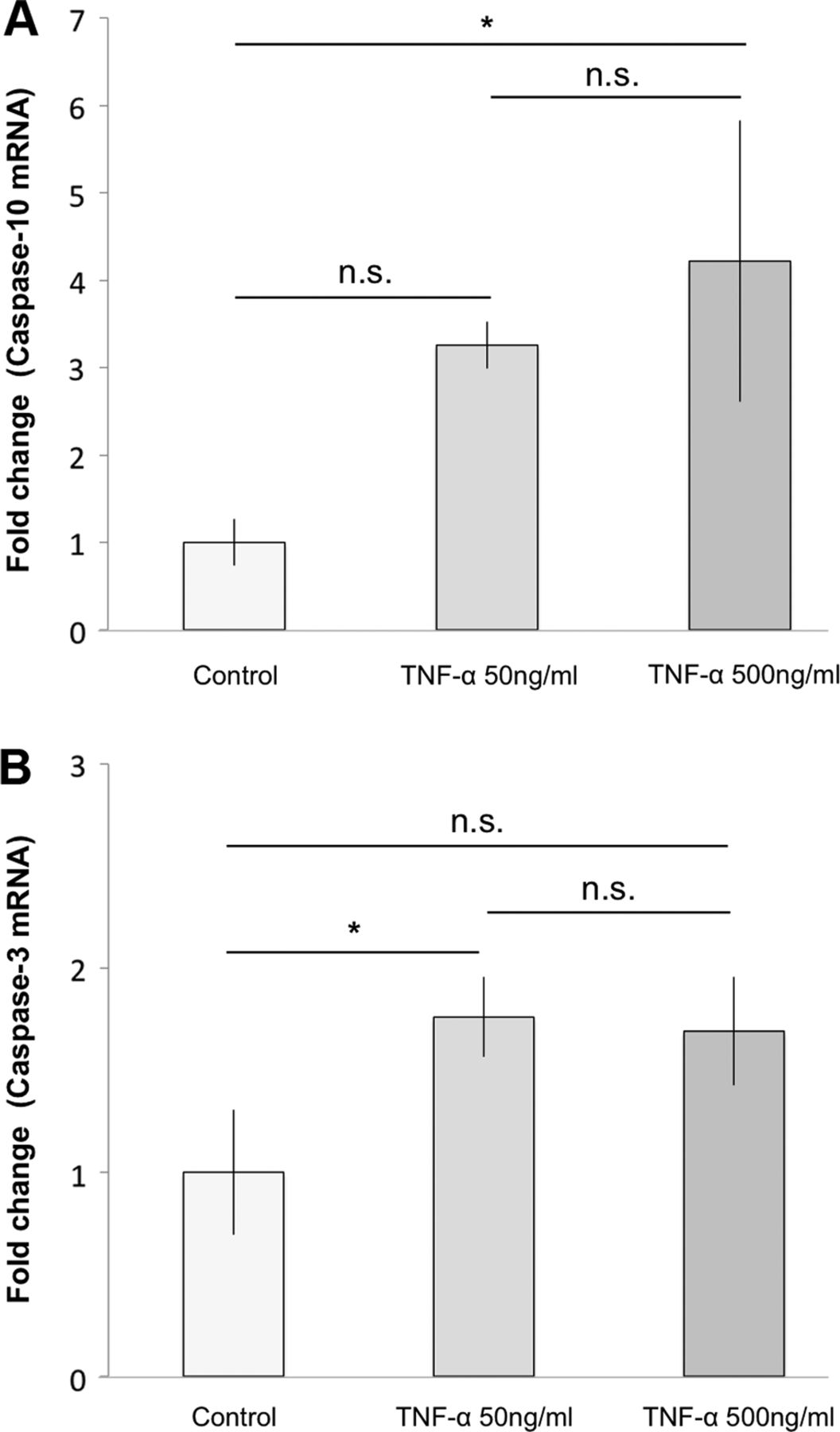
Gene expression results for caspase-10 mRNA showed a significant increase for cells treated with 500 ng/mL of TNF-α for 7 h compared to the control (4.2-fold change; p=0.02; figure 4A). Cells treated with only 50 ng/mL of TNF-α showed an increase as compared to control after 7 h of exposure, although this was not statistically significant (p=0.08; figure 4A). Gene expression results for caspase-3 mRNA showed a significant increase for cells treated with 50 ng/mL of TNF-α compared to the control (1.8-fold change; p=0.04; figure 4B), after 7 h of exposure. The increase seen for cells treated with 500 ng/mL of TNF-α was close to significant (p=0.05; figure 4B).

Effects of tumour necrosis factor-α (TNF-α) on the apoptotic signalling pathway at mRNA level. Subjecting cultured human tenocytes to TNF-α for 7 h significantly increased main parameters of the apoptotic pathway. (A) 500 ng/mL of TNF-α significantly increased caspase-10 mRNA as compared to the control (4.2-fold change; *p<0.05). 50 ng/mL also showed an increase compared to control, although this was not statistically significant (n.s) (p=0.08). (B) In total, 50 ng/mL of TNF-α significantly increased caspase-3 mRNA in comparison to the control (1.8-fold change; *p<0.05). The increase seen for cells treated with 500 ng/mL of TNF-α was not statistically significant (n.s.), although close to significant (p=0.05). Error bars indicate the SD.
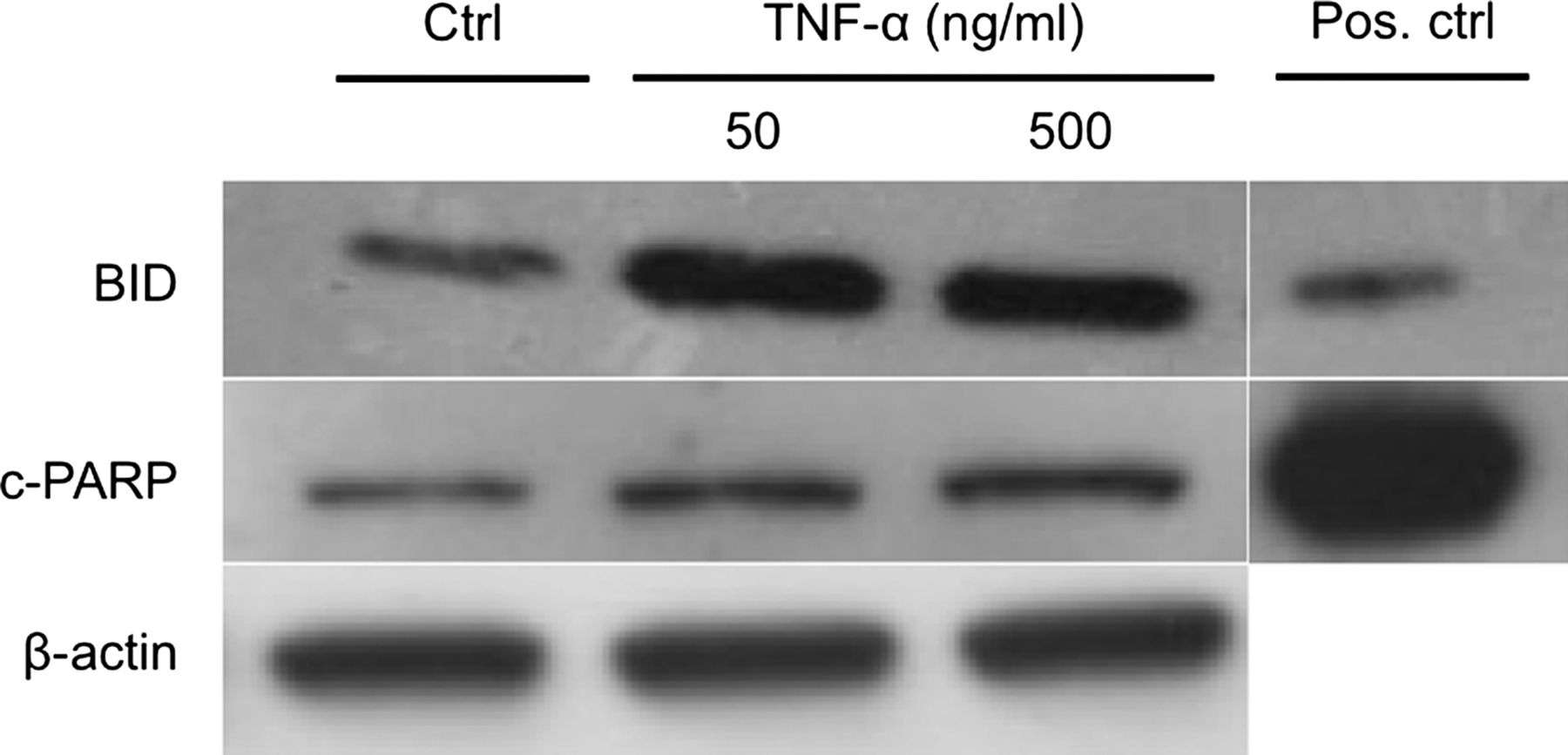
To determine BID and c-PARP, cells were treated with TNF-α for 12 h. On protein level both c-PARP and BID showed an increase after 12 h of exposure to both 50 and 500 ng/mL of TNF-α (figure 5). However, the increase was more obvious for BID (figure 5).

Effects of tumour necrosis factor-α (TNF-α) on the apoptotic signalling pathway at protein level. Twelve hours of exposure to TNF-α resulted in an increase of both BID and cleaved-PARP (c-PARP) in cultured human tenocytes. A more obvious increase in comparison to the control (Ctrl) was seen for BID, as compared to c-PARP, after treatment with TNF-α. β-Actin was used as a loading control. Jurkat apoptosis cell lysate was used as a positive control (Pos. ctrl).
SP reduces TNF-α-induced cell death
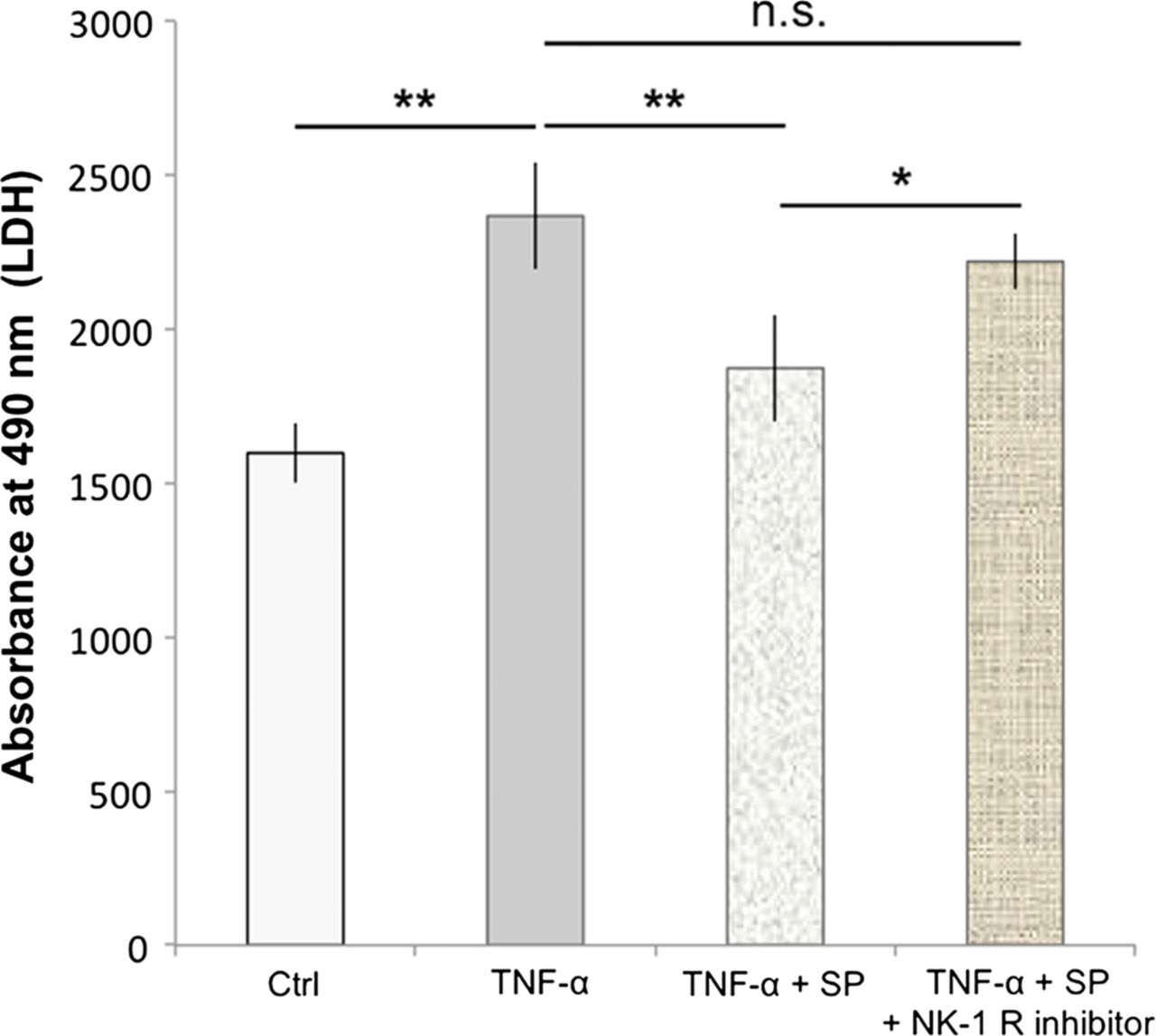
Cells treated with 500 ng/mL of TNF-α for 48 h showed a significant increase in the amount of lactate dehydrogenase (LDH) release compared to the control (p<0.01; figure 6). Pretreatment with SP at concentrations of 10−7 M significantly reduced the TNF-α-induced LDH release, as compared to TNF-α alone (p<0.01; figure 6). This effect of SP was inhibited by the use of the NK-1 R inhibitor (10−6 M).
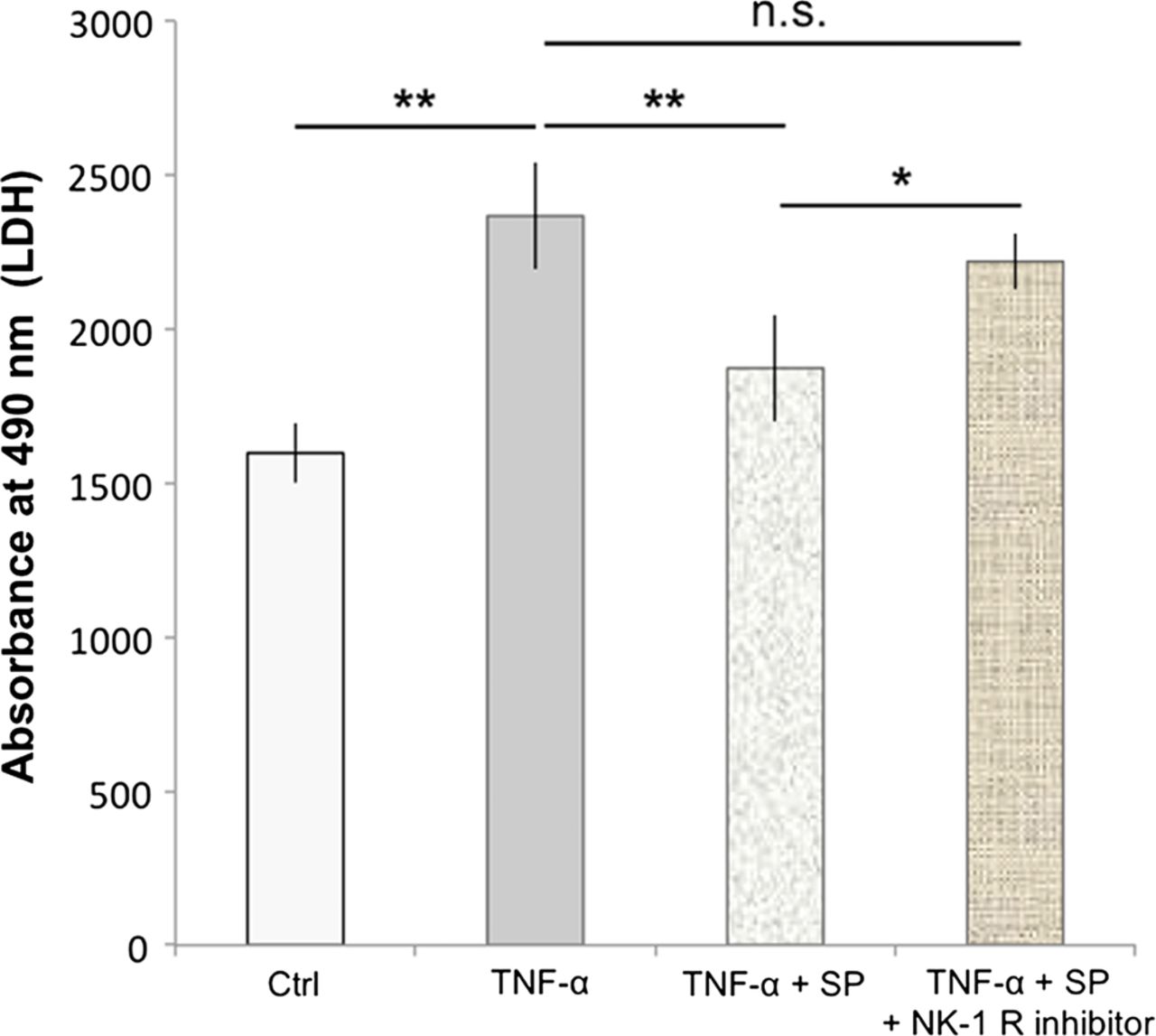
Substance P (SP) reduces TNF-α-induced cell death. Human cultured tenocytes treated with 500 ng/mL of TNF-α for 48 h showed a significant increase in the amount of lactate dehydrogenase (LDH)-release compared to the control (Ctrl). Pretreatment with SP at concentrations of 10−7 M significantly reduced the TNF-α-induced LDH-release, as compared to TNF-α alone. However, when the neurokinin-1 receptor (NK-1 R) inhibitor (at concentrations of 10−6 M) was added to the SP and TNF-α treatments of the cells, the LDH release was not significantly (n.s.) different from that after incubation with TNF-α alone, but significantly increased as compared to incubation with TNF-α together with SP. Thus, the SP effect of reducing the TNF-α-induced LDH-release, that is, cell death, was effectively inhibited by blocking the NK-1 R, showing that the SP effect is mediated through this receptor. Error bars indicate SD (*p<0.05; **p<0.01).
SP reduces TNF-α-induced increase in c-PARP and BID
When added prior to 500 ng/mL of TNF-α, SP decreased the amount of c-PARP and BID as compared to TNF-α alone (figure 7). The effects of SP were blocked when incubated simultaneously with the NK-1 R blocker (figure 7).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Substance P (SP) reduces the tumour necrosis factor-α (TNF-α)-induced apoptosis in human tenocytes. The increase in expression of BID and cleaved-PARP seen in cultured human tenocytes after incubation with TNF-α as compared to control (Ctrl), was clearly reduced when cells were pretreated with SP. Blocking of the SP receptor with an NK-1 R inhibitor (NK-1 R inhib.) reduced this effect of SP. β-actin was used as a loading control and jurkat apoptosis cell lysate was used as a positive control (Pos. ctrl; 12 h of incubations).
Discussion
In this study, using an in vitro model, we showed that human Achilles tendon cells (tenocytes) from healthy donors express both of the known TNF-α receptors; TNF-R1 and TNF-R2. We furthermore demonstrated that TNF-α decreases cell viability and activates key mediators of the apoptotic pathway, such as caspase-10, caspase-3, BID and PARP, in the tenocytes. Moreover, TNF-α increased cell death in the tenocyte cultures, as seen by augmented LDH release. However, when cells were treated with TNF-α together with SP, the increase in LDH release was inhibited, as was the activation of BID and PARP. Hence, our results demonstrate that SP has the ability to prevent the apoptotic effects of TNF-α in human tenocytes.
TNF-α induces apoptosis in human tenocytes
Our study provides information that TNF-α induces apoptosis in human tenocytes. However, the effects were most clearly seen early in the apoptotic cascade, as we observed a more than fourfold increase in caspase-10 mRNA, as well as a distinctly increased amount of BID. The relatively minor increase noticed for the late stage mediators (caspase-3 mRNA and c-PARP) suggests that other factors, possibly inhibiting later stages of apoptosis, are involved. It is known that TNF-α has the ability to activate both its receptors simultaneously, thus stimulating signals of apoptosis (via TNF-R1) and cell survival (via TNF-R2) at the same time. The balance between the two signals ultimately determines the amount of apoptosis.24 This dual effect of TNF-α, and the fact that both receptors are here shown to be present on the tenocytes, albeit TNF-R1 more distinctively, might explain why we did not observe a more potent negative effect of TNF-α in our results from the cell viability assay. The dual effect of TNF-α might also explain the higher increase of casapase-3 mRNA after tenocytes were subjected to 50 ng/mL of TNF-α as compared to 500 ng/mL (although statistically not significant). The cell survival induced by TNF-R2 stimulation might require higher concentrations of TNF-α to become activated, because the receptor is less expressed and consequently it might reduce the effect of TNF-α regarding caspase-3 mRNA increase (which possibly is induced via TNF-R1) only at higher concentrations of TNF-α. The current study does not elucidate the entire signalling pathway of TNF-α at play in the tenocytes. However, since TNF-α has the ability to mediate either proapoptotic effects via TNF-R1 or antiapoptotic effects via TNF-R2, it would be of interest to perform receptor blocking of the TNF-α receptors in future studies, to fully evaluate the effects of TNF-α in tenocytes. Nevertheless, our study showed that TNF-R1 is more intensely expressed in tenocytes than TNF-R2, possibly facilitating an overweight for the proapoptotic TNF-α stimulus, which would also be in accordance with the outcome of the TNF-α stimulation of our cells. The results here discussed, that is, the relatively minor increase in late stage apoptosis mediators (caspase-3 mRNA and c-PARP), as compared to the early mediators (caspase-10 mRNA and BID), might also be explained by the time-points used for analysis. It is possible that analysis at later time-points would result in an increase of the late apoptotic mediators.
In light of the possible dual effect of TNF-α on tenocytes discussed, it is of further interest to note that we have also found a significant increase in NF-κB activation after 4 h of treatment with TNF-α (preliminary results; data not shown). It is known that in response to stimuli (such as that brought on by TNF-α), NF-κB translocates from cytoplasm to nucleus, ultimately resulting in increased cell survival.31 ,32 Hence, the relatively minor activation of the late stages apoptotic regulators caspase-3 and PARP in response to TNF-α, in the current study, might imply that TNF-α activation of NF-κB mediates cell survival also in human tenocytes via inhibition of mechanisms further downstream of regulators such as caspase-10 and BID in the apoptotic cascade.
The possible role of TNF-α-induced apoptosis in tendinosis, and of SP as an antiapoptotic agent
It has previously been shown that SP and its preferred receptor NK-1 R as well as TNF-α together with its receptors, TNF-R1 and TNF-R2, are expressed in human tenocytes in both healthy and tendinosis tissue.9 ,19
The exact functions of these substances and their receptors have previously not been elucidated. However, it is possible that tenocytes inside the tendon, expressing TNF-R1 and TNF-R2, are stimulated by TNF-α released by the immune cells of the paratenon,20 that is, the loose connective tissue outside the tendon and/or by TNF-α produced by both the tenocytes themselves. Furthermore, it has been shown that TNF-R1 is significantly more expressed in tenocytes in Achilles tendinosis than in healthy controls.19 Since TNF-R1 is known to mediate apoptotic effects it is possible that the TNF-α signalling, whether originating from tenocytes and/or immune cells, is causing the excessive amount of apoptosis seen in tendinosis16 via stimulation of the TNF-R1 of the tenocytes. The findings of the current study have provided evidence that TNF-α indeed has the ability to induce apoptosis in human tenocytes. We have also shown that SP has the ability to reduce the apoptotic effects of TNF-α via stimulation of the NK-1 R. It is possible that SP may be contributing to the hypercellularity that is observed in tendinosis, both via stimulating proliferation of tenocytes, as shown in one study,11 and via inhibition of apoptosis in these cells, an effect shown in the current study but also in a recent study after anti-Fas-induced apoptosis.28 Hypothetically, TNF-α-induced apoptosis in chronic stages of tendinosis might be a way for the tendon to try to limit an overabundance of collagen-synthesising cells, and thereby constricting excessive collagen production. If so, it is possible that SP, via NK-1 R, plays an important role in the development of tendinosis, as its antiapoptotic effects may be contributing to the hypercellularity and ultimately excessive collagen depositions.
Conclusions
In conclusion, the current study demonstrates an apoptotic effect of TNF-α on tenocytes and also that SP can reduce this effect via NK-1 R stimulation. Considering that SP has previously been shown to increase significantly in tendinosis as a response to overload10 ,11 and to stimulate proliferation of tenocytes,11 ,12 this study confirms SP as a potent regulator of cell turnover and a possible contributory factor to the hypercellularity seen in tendinosis. The antiapoptotic effect of SP might hypothetically hinder an important way for the tendon to self-limit the amount of collagen-producing cells that contributes to excessive and faulty collagen deposition in tendinosis. Based on future studies further exploring these hypotheses, blocking of NK-1 R as a way of interfering with the possible deleterious effects of SP could be considered in experimental treatments of tendinosis.
What are the new findings?
-
Tumour necrosis factor-α (TNF-α), known to be produced by tendon cells (tenocytes) and immune cells of the paratenon in tendinosis, induces apoptosis in human tenocytes in vitro.
-
Substance P (SP), known to be up-regulated in tendons/tenocytes in response to stress, mediates an antiapoptotic effect, via the neurokinin-1(NK-1) receptor, in TNF-α-induced apoptosis of human tenocytes and may thus play an important role in the development of tendinosis, as its antiapoptotic effect could be contributing to the tenocyte hypercellularity and ultimately excessive collagen depositions seen in tendinosis.
How might it impact on clinical practice in the near future?
-
Blocking of the neurokinin-1 receptor, as a way of interfering with the possible deleterious effects of SP, should be considered in experimental treatments of tendinosis in the future.
Acknowledgments
The authors would like to thank Dr Sandrine Le Roux and Mr Peter Boman for scientific advice and technical help, and Dr Gustav Andersson for skilful scientific artwork (figure 1). The authors also extend their deepest appreciation to professor Håkan Alfredson for surgically providing the tissue material and to Mrs Lotta Alfredson for coordinating donations.
References
Footnotes
-
Contributors LJB and PD were responsible for the conception and design of the study. HA collected tendon biopsies. LA coordinated donations. GA made the scientific drawing in figure 1. LJB and DEE performed the experiments. LJB, PD and DEE analysed the data. LJB, DEE and PD wrote the manuscript. All authors had full access to all the data and contributed to the interpretation of the findings and critical revision of the manuscript. PD and LJB are the guarantors.
-
Funding This work was primarily supported by a grant from the national Swedish Research Council (521-2009-2921 to PD) and an Umeå University Young Researcher Award (to PD). The project was furthermore supported by grants from the Swedish National Centre for Research in Sports (P2011-0170 to PD) and the Swedish Society of Medicine (SLS-176511 and SLS-248321 to PD). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
-
Competing interests None.
-
Ethics approval The collection of human tendon samples was approved by the Regional Ethical Review Board in Umeå (04-157M).
-
Provenance and peer review Not commissioned; externally peer reviewed.